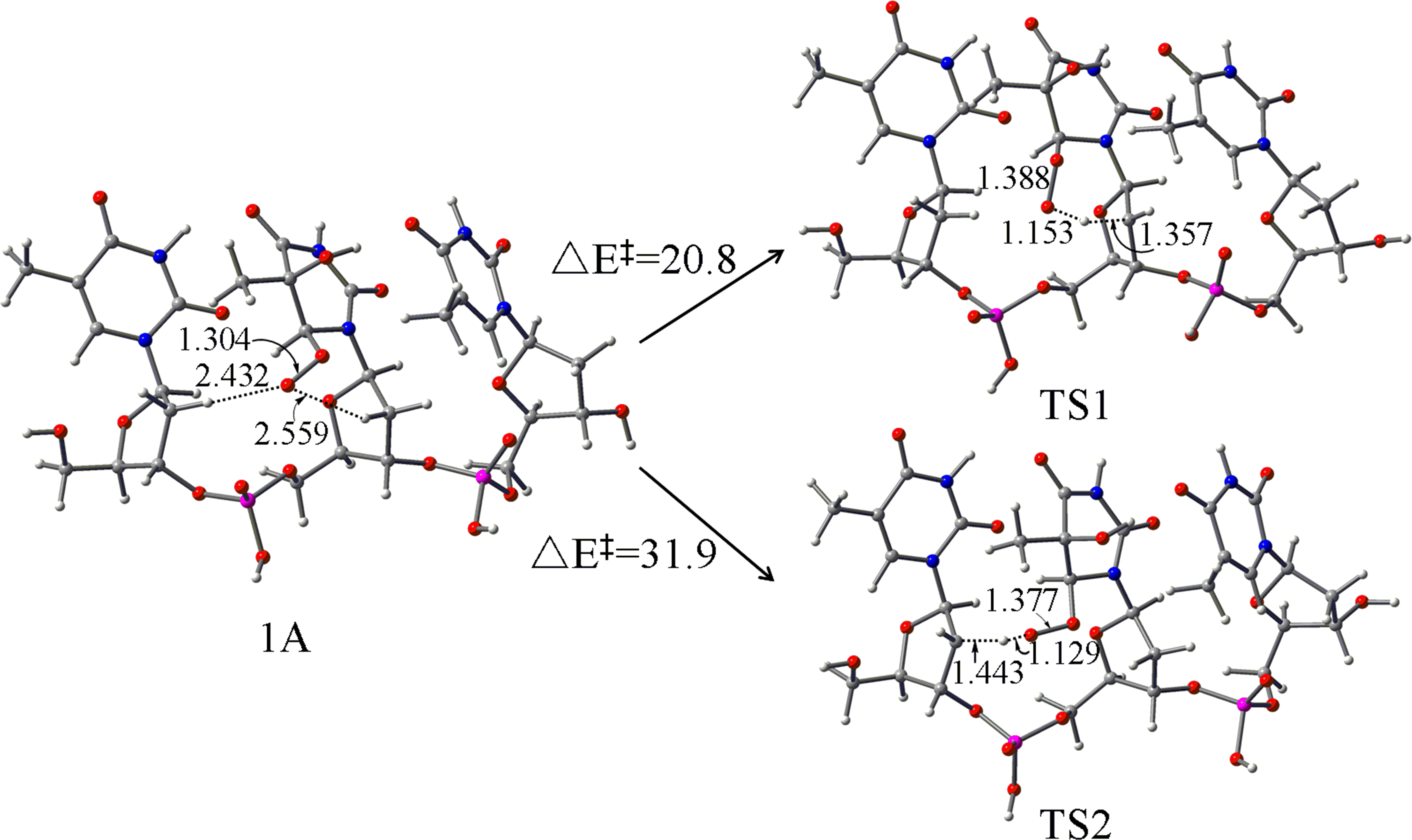
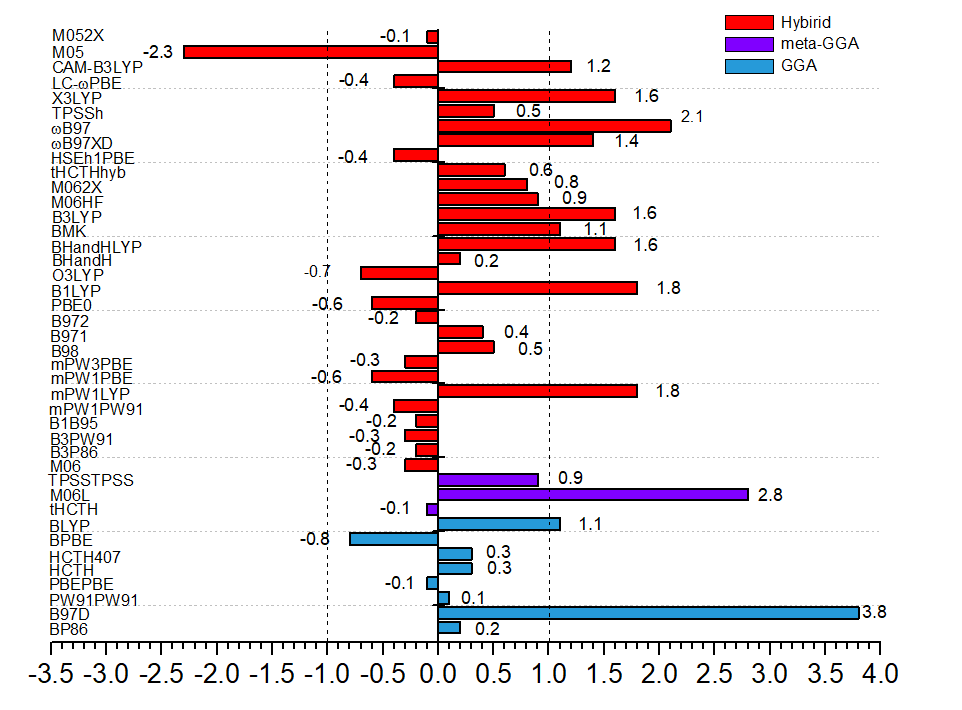
The performance of a series of density functionals has been tested for the insertions of ethylene, methyl acrylate (MA), and vinyl bromide (VB) catalyzed by α-diimine palladium complexes. Sixty-seven density functionals are screened, and the results are compared with available experimental data. Eleven hybrid functionals (M06, BHandH, mPW1PW91, HSEh1PBE, mPW3PBE, LC-ωPBE, mPW1PBE, PBE0, M06-HF, M06-2X, M05-2X) showed better performance in the insertions of both ethylene and MA, and could be therefore suitable for ethylene-MA copolymerization. Meanwhile, three GGA (PW91PW91, HCTH, HCTH407), two meta-GGA (TPSSTPSS, tHCTH), and ten hybrid functionals (M06, BHandH, TPSSh, B971, B98, B1B95, PBE0, M06-2X, tHCTHhyb, M05-2X) perform well in the ethylene-VB copolymerization. Besides, nine D3 or D3BJ augmented functionals are found to be suitable for both copolymerization systems. The D2 dispersion correction overestimated insertion energy barriers of these monomers and is unsuitable for such copolymerization. In addition, the double-zeta basis set is found to be sufficient for solvation single-point calculation of these systems.