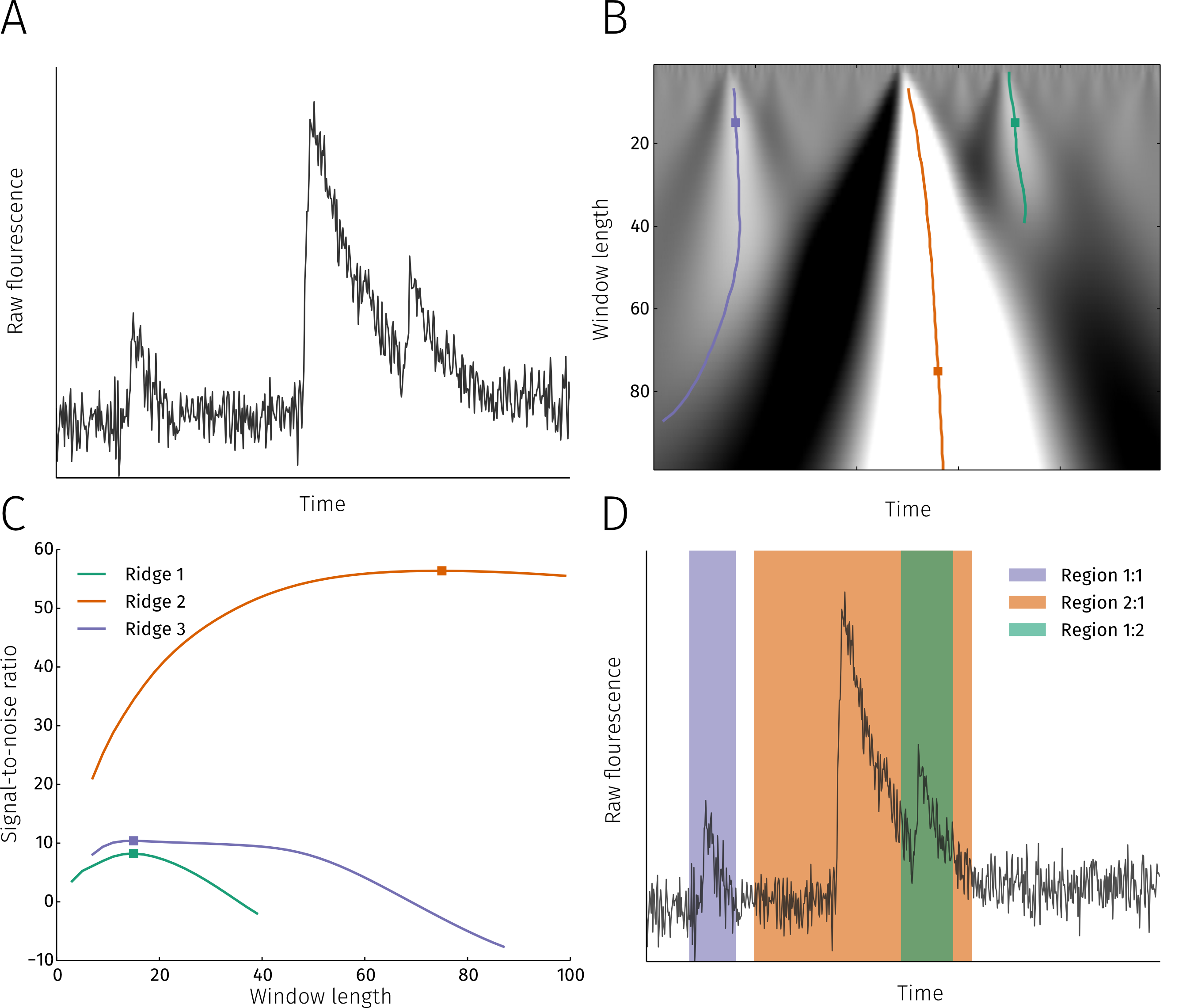
Analysis of Ca2+ signaling in cardiac cells is always a trade-off between acquisition speed and signal-to-noise ratio. This becomes especially apparent in confocal microscopy, during fast 2D scanning or when recording fluorescence signals from the sarcoplasmic reticulum, for example. Methods have been developed to remedy this via 'denoising' the image by fitting each pixel with a transient function. So far, adoption of such methods has been hindered by a number of limitations (e.g., inability to fit local, concurrent and consecutive events) and the limited availability of a customizable implementation. Here we present a novel method for performing per-pixel denoising of confocal frame- and linescans. Our algorithm permits the extraction of spatiotemporally overlapping events (e.g., a Ca2+ spark occurring during the decaying phase of a Ca2+ wave) and is able to detect various different types of events within a pixel time course. The method estimates a non-constant baseline for each pixel, negating the necessity of using background regions or self-ratio methods prior to performing the analysis. Furthermore, by applying a clustering algorithm, detected single-pixel events are grouped into physiologically relevant events spanning multiple pixels (sparks, waves, puffs,transients, etc.), from which traditional parameters such as FDHM, FWHM, amplitude, wave speed, rise and decay times, can be easily extracted. The method has been implemented as a cross-platform open source software with a comprehensive and easy to use graphical user interface. We have applied our method to analyzing linescans of repetitive Ca2+ sparks from individual RyR clusters in isolated ventricular cardiomyocytes; high-speed (150 frames/sec) framescans containing alterations in Ca2+ release events in atrial myocytes; and parallel analysis of Ca2+ release dynamics in the sarcoplasmic reticulum and cytosol.