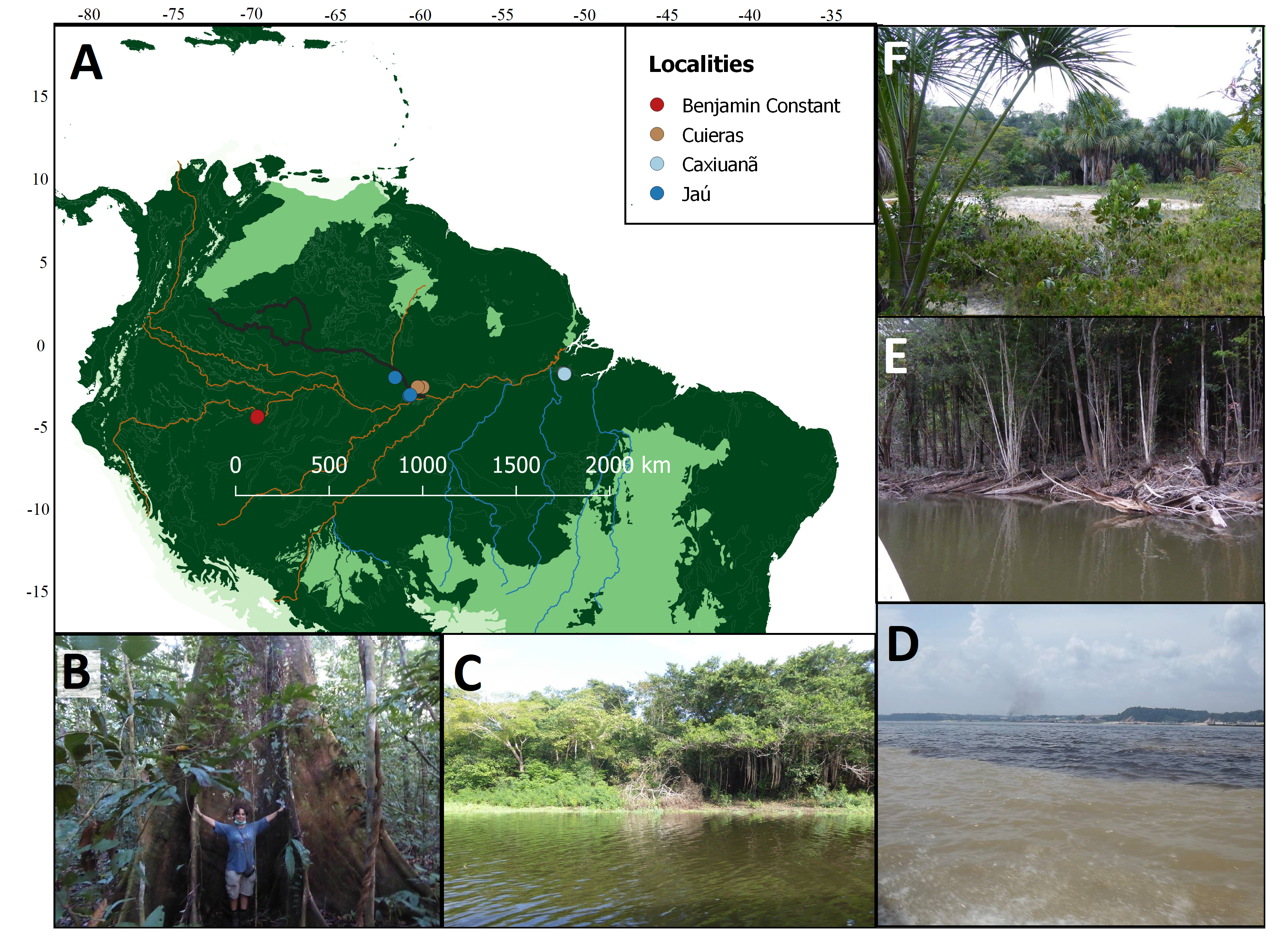
Over the last few centuries, millions of plant specimens have accumulated within herbaria and biocultural collections. These include type specimens, used to define species, and populations that are rare, extinct or difficult to access. They therefore represent a considerable resource for a broad range of scientific uses. However, collections degrade over time, and become increasingly difficult to characterise their genetic signatures, even considering exponential advancements in sequencing technologies. Here, we tested the genotyping performance on highly degraded samples using a commonly used high-throughput sequencing (HtS) technique, genome skimming, against a recent alternative target capture kit, the universal set Angiosperm-353. We performed phylogenomic analyses of modern leaf and historical barks of Cinchona, including 23 historical barks and six fresh leaf specimens. DNA within historical barks is highly degraded, therefore a customised DNA extraction method was developed before library preparation. We show that sample degradation over time directly impacted the quantity and quality of the data produced by both methodologies (in terms of reads mapped to the references). However, we find that both approaches generate enough data to infer phylogenetic relationships, even between highly degraded specimens that are over 230 years old. Within historical barks, the target capture kit is more advantageous than genome skimming in profiling Cinchona species since it was possible to retrieve nuclear and plastid data to infer phylogenies. This study showcases the value of historical samples in genetic studies and paves the way for further experiments across different taxonomic groups with varying levels of genetic variation or hybridisation.