AUTHOREA
Log in
Sign Up
Browse Preprints
LOG IN
SIGN UP
Mahmoud Abdelnabi
Public Documents
4
August 11, 2020
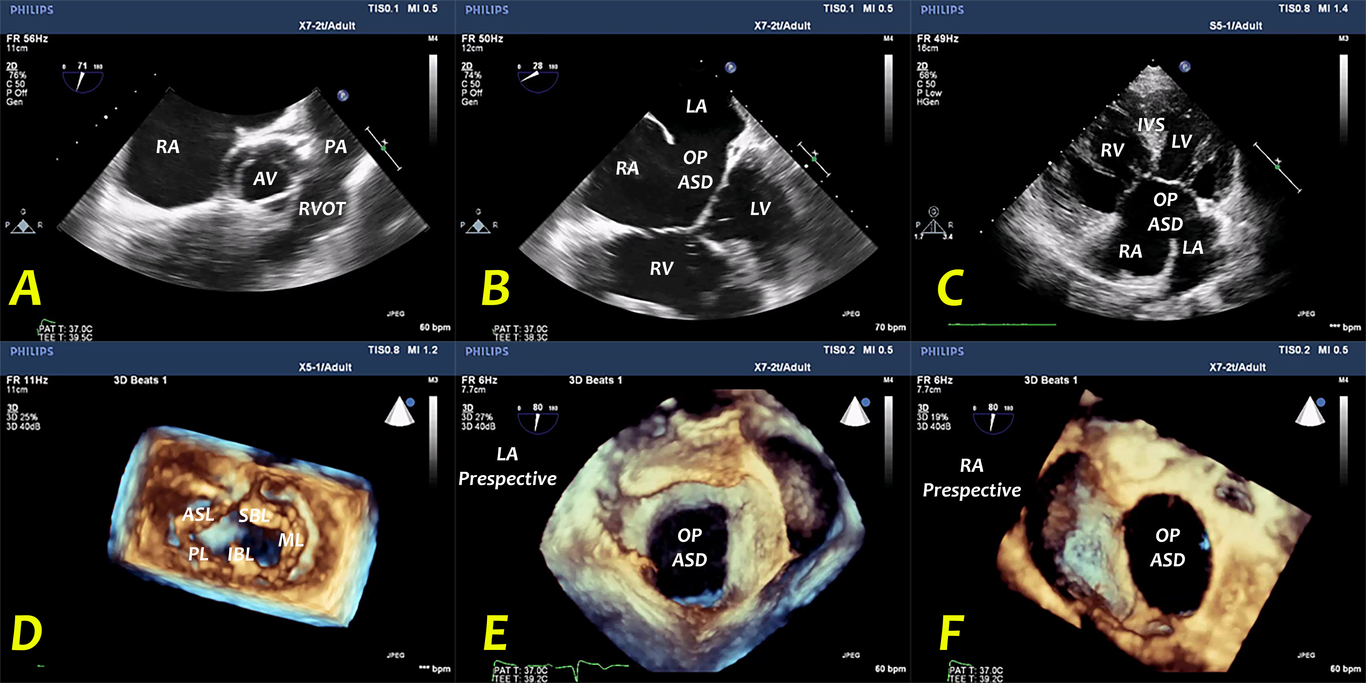
Imaging of a Case of Atrioventricular Septal Defect: The added value of using the Thi...
Hoda Shehata, Mahmoud Abdelnabi, Abdallah Almaghraby, et al.
June 18, 2020
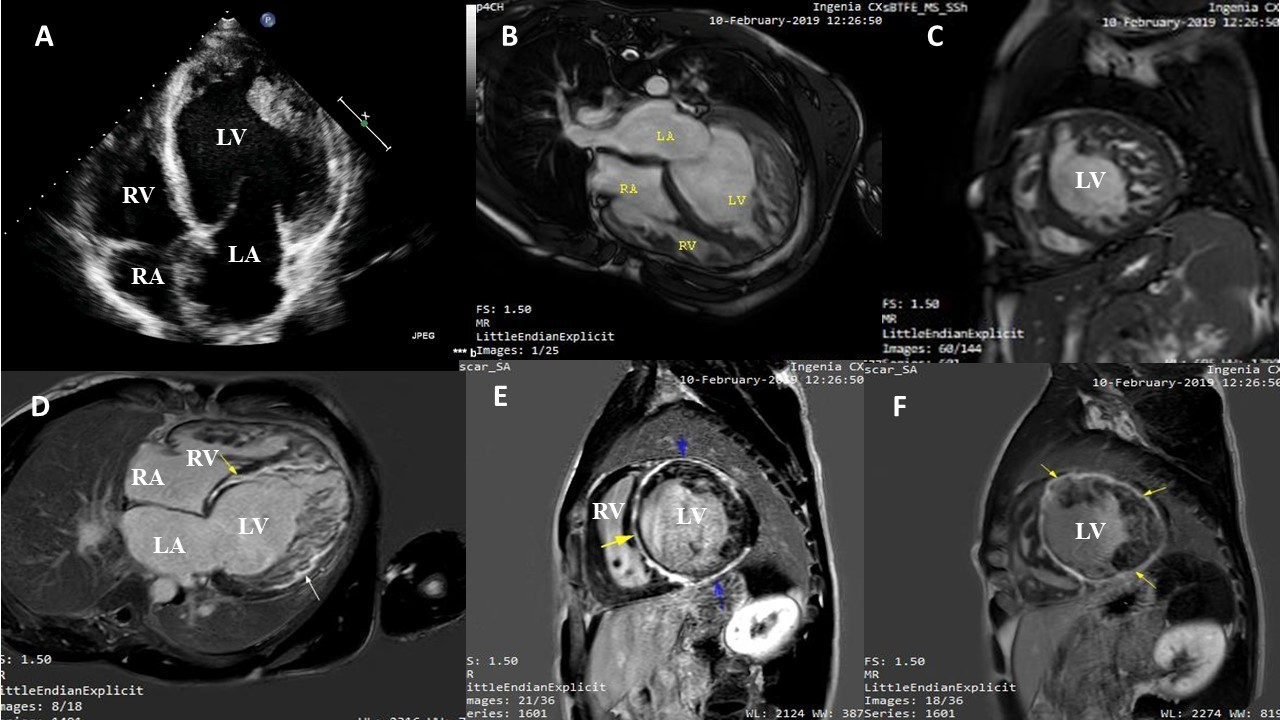
An unusual pattern of late gadolinium enhancement (LGE) in a bi-ventricular non-compa...
Fatma Elkafrawy, Mahmoud Abdelnabi, Abdelraman Assal, et al.
April 10, 2020
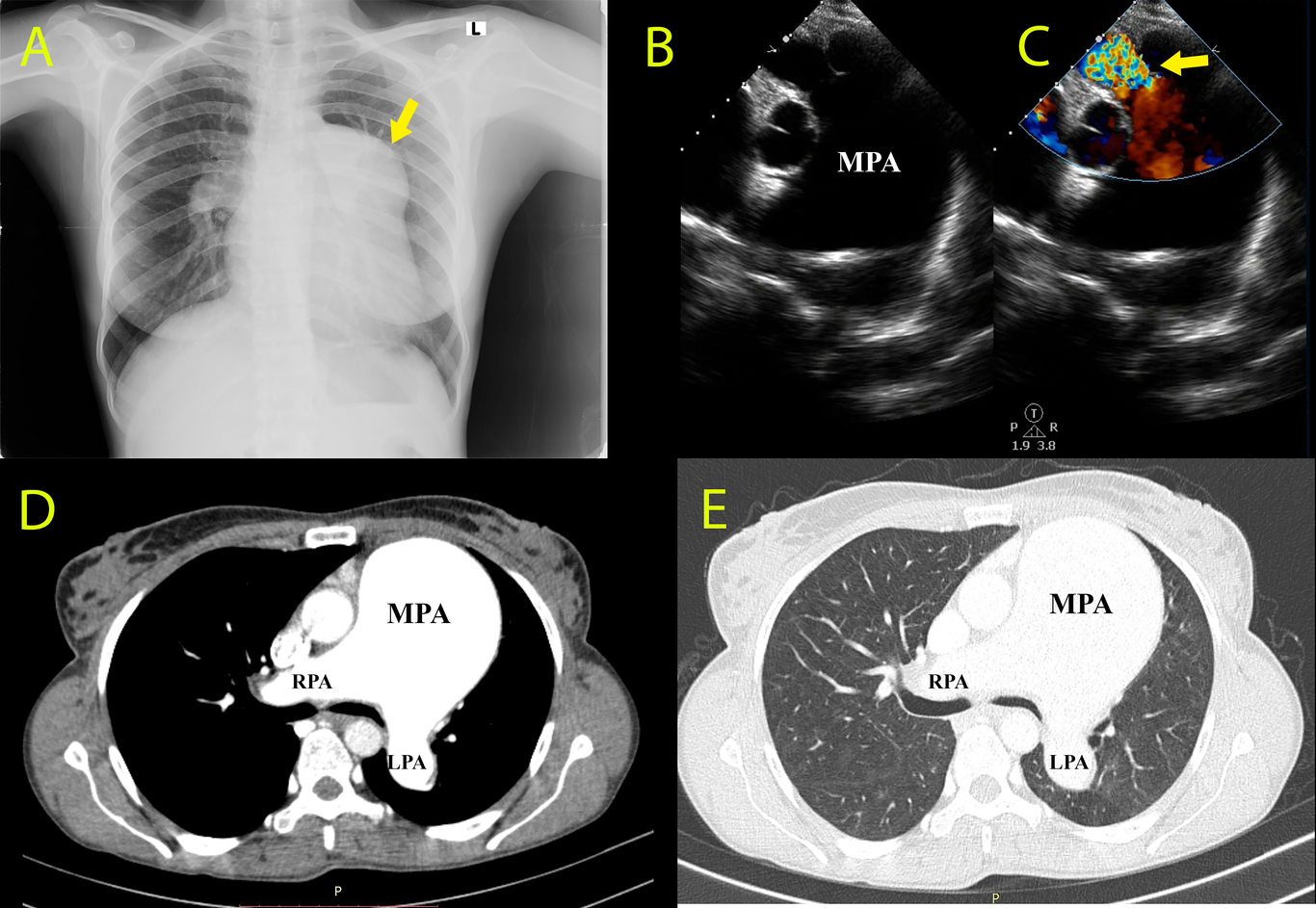
Ortner's syndrome due to large bilharzial pulmonary artery aneurysm
Mahmoud Abdelnabi, Nouran Eshak, Yehia Saleh, et al.
April 09, 2020
A Long-term unrepaired Fallot tetralogy survivor treated only with a Classical Blaloc...
Mahmoud Abdelnabi, Hoda Shehata, Fatma Elkafrawy, et al.


/6 in 1 with labels (1).png?1586646173)