AUTHOREA
Log in
Sign Up
Browse Preprints
LOG IN
SIGN UP
Melissa Hawkins
Public Documents
2
April 22, 2020
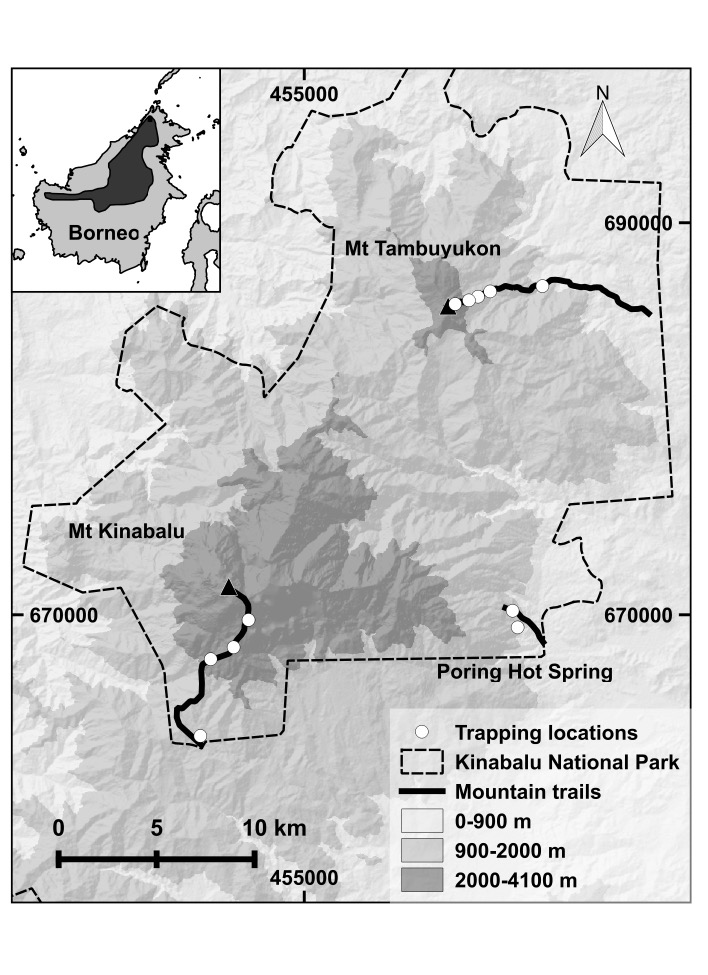
High gene flow across heterogeneous tropical montane environments in a Bornean endemi...
Lillian Parker, Melissa Hawkins, Miguel Camacho-Sanchez, et al.
May 27, 2020
Assessing the levels of microsatellite allelic dropout in museum specimens using high...
Stella Yuan, Eric Malekos, Melissa Hawkins, et al.