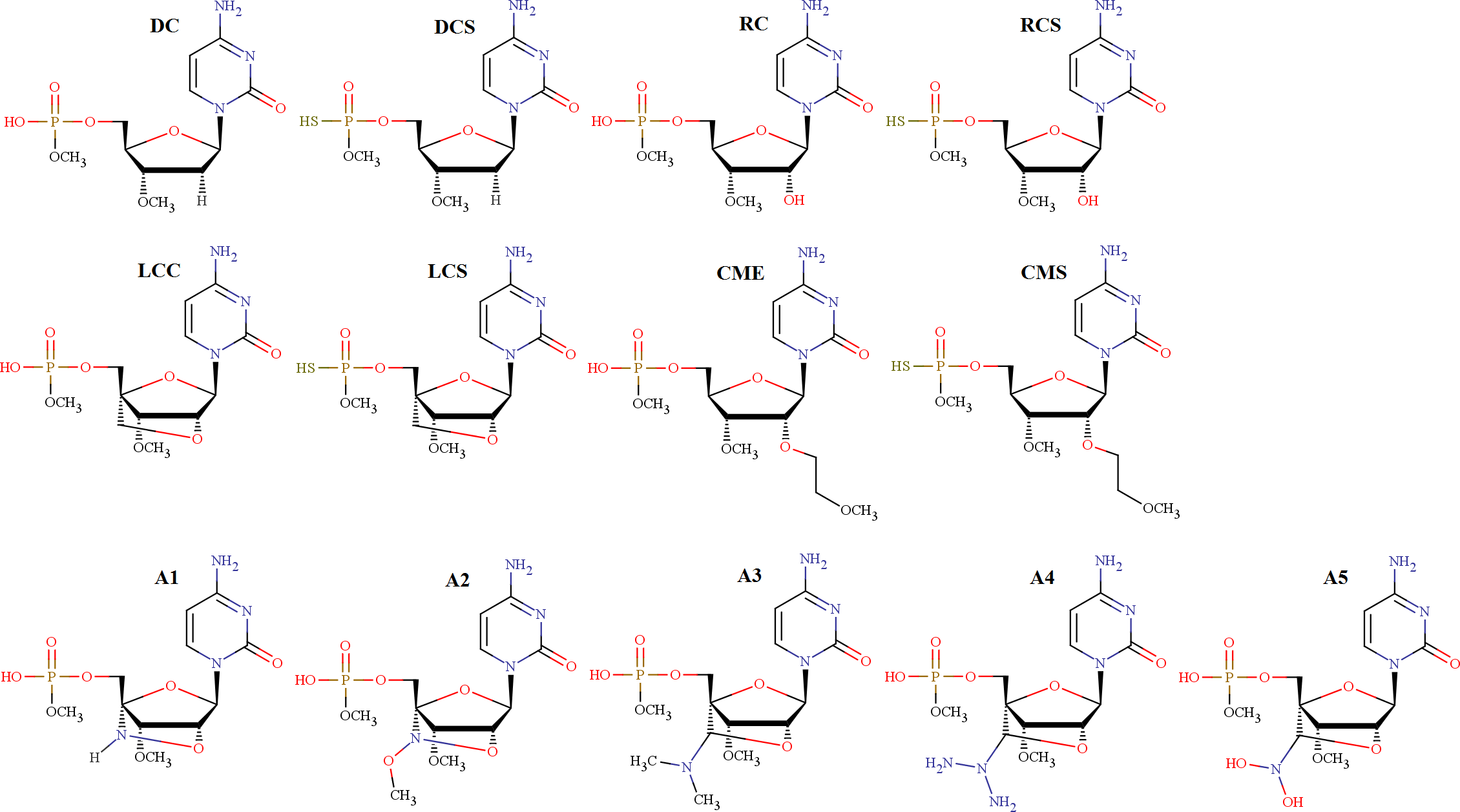
Antisense technology has been developed as the next generation drug discovery methodology by which unwanted gene expression can be inhibited by targeting mRNA specifically with antisense oligonucleotides. The computational studies of antisense modifications based on phosphorothioate (PS), methoxyethyl (MOE), locked nucleic acids (LNA) may help to design better novel modifications. In the present study, five novel LNA based modifications have been proposed. The conformational search has been done to identify the most stable and alternative stable conformations. The geometry optimization followed by single point energy calculation has been done at B3LYP/6-31G(d,p) level for gas phase and B3LYP/6-311G(d,p) level for the solvent phase for all modifications at monomer as well as base pair level. The electronic properties and the quantum chemical descriptors of the antisense modifications were derived and compared. The local and global reactivity descriptors, such as hardness, chemical potential, electronegativity, electrophilicity index, Fukui function all were calculated at the DFT level for the optimized geometries. A comparison of global reactivity descriptors confirmed that LNA based modifications are the most reactive modifications. They may form a stable duplex when bound to complementary nucleotides, compared to other modifications. Therefore, we are proposing that one of our proposed antisense modifications (A3) may show strong binding to the complementary nucleotide as LNA and may also show reduced toxic effects like MOE. The base pair studies may help us to understand the extent to which our proposed modifications can form standard Watson-Crick base pairing required for oligomer duplexes. This computational approach may be very useful to propose novel modifications prior to undergoing synthesis experimentally in the area of antisense or RNAi technology.