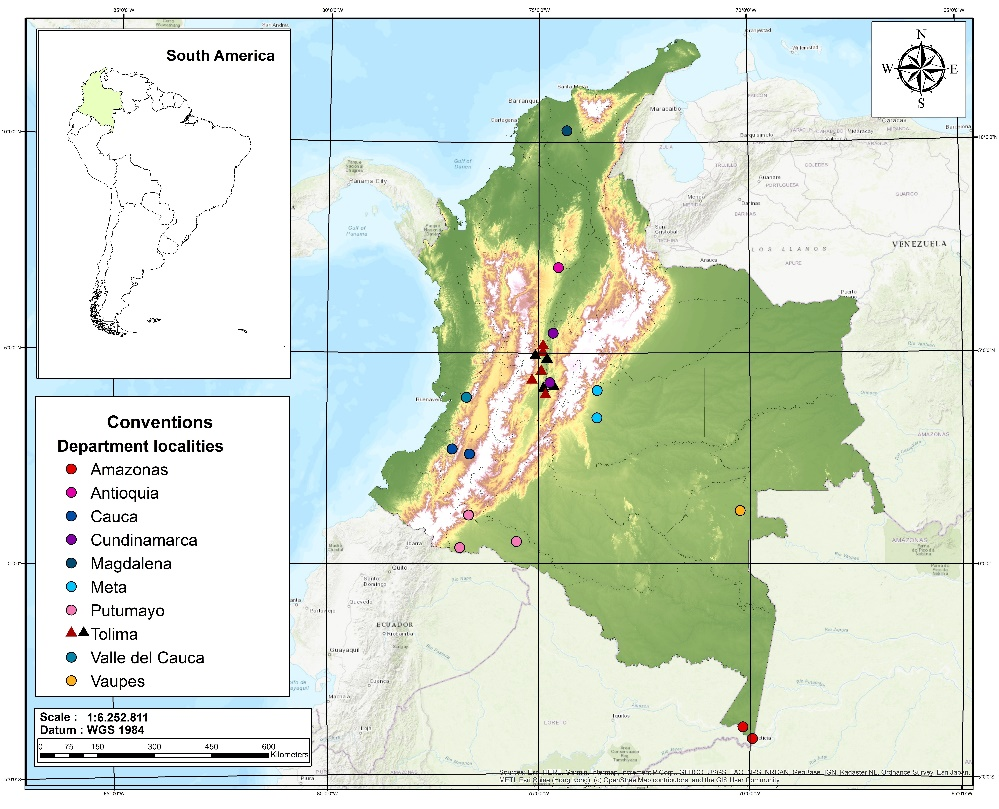
1. Phyllostomid bats exhibit great diversity in skull size and morphology that reflects the degree of resource division and ecological overlap in the group. In particular, Stenodermatinae has high morphological diversification associated with cranial and mandibular traits that is associated with the ability to consume the full range of available fruits (soft and hard). In terms of morphology, performance (bite force) appears to play an important role in niche partitioning among bat species, however, very few studies have confirmed these relationships using functional cranial traits. 2. Here, we analyzed craniodental traits and their relationship to the bite force in 308 specimens distributed in seven species of stenodermatine bats with two foraging types: nomadic and sedentary frugivorous bats. We evaluated 19 functional traits of the skull and jaw related to feeding and bite force in live animals by correcting bite force with body size. We used a GLM model and post hoc tests to determine possible relationships and differences between cranial traits, species, and sex. 3. The results showed that there is significant interspecific variation between stenodermatines that are nomadic and sedentary. The greatest variation in bite force within species was explained by the mandibular toothrow length (MANDL) between sexes, which was greater in females. The larger species of Artibeus, together with Platyrrhinus helleri, Uroderma convexum and Sturnira giannae, which have a greater length of the skull, condylo-incisor, condylo-canine, mandibular toothrow and height of the coronoid, exhibit greater bite force. By contrast, the smaller species A. anderseni and A. phaeotis have short skulls and the lowest values of bite force, which suggests that the size of the skull confers a biomechanical advantage. 4. Our results highlight the usefulness of analyzing functional traits related to feeding to establish the performance of bats in terms of the bite force.