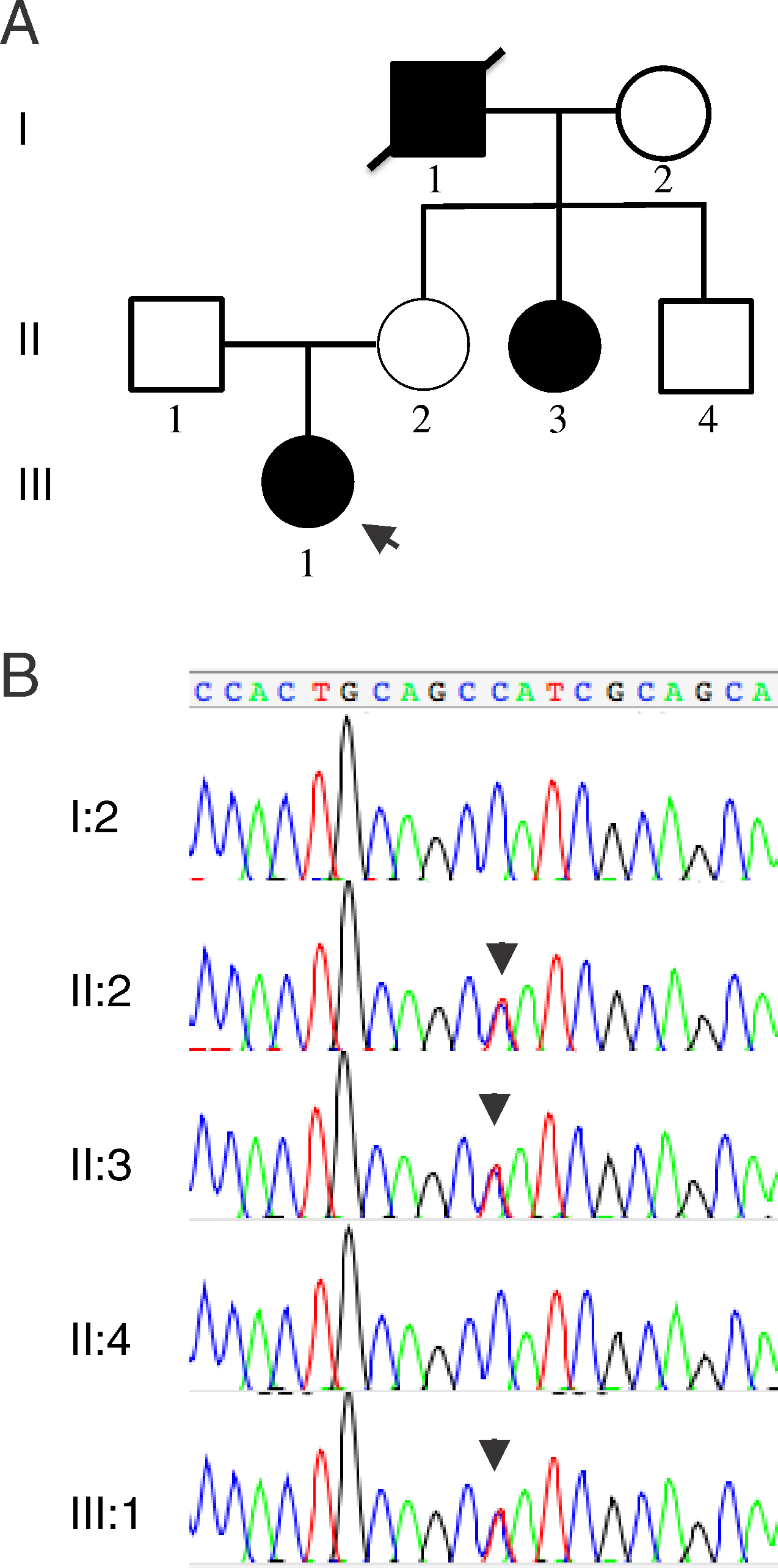
BackgroundSystemic sclerosis, also known as SSc or scleroderma, is an autoimmune disease characterized by a triad of microvascular dysfunction, immune dysfunction, and generalized fibrosis in connective tissues and organs [1]. One of the most concerning aspects of the disease is that mortality has not improved greatly over the last several decades because there is a critical lack of therapies to address the fibrotic process [2]. The urgent need for innovation in SSc is one of the motivations of the genetics community in attempting to explore the hereditary underpinnings of this condition. Genetic epidemiology has shown convincing evidence of familial aggregation, with increased risk to siblings and first degree relatives as well as substantial epidemiologic overlap with other autoimmune diseases [3]. The etiology of the disease is multifactorial, with poorly-understood environmental influences and a complex mode of genetic inheritance. Since SSc is a relatively rare disease, most cases appear sporadically, without family history [3]. Recent advances in genomic technology, such as high-density genotyping on microarrays, have made possible genome-wide association studies (GWAS) that have enhanced the genetic understanding of SSc.The single stand-out genetic risk for SSc is associated with an array of variants in the major histocompatibility complex (MHC), containing the human leukocyte antigen (HLA) genes [4], a pattern seen in a wide array of autoimmune diseases. The first large GWAS revealed associations with non-coding SNPs at a number of loci in addition to the HLA, including IRF5, STAT4, CD247, CDH7, and IRF4 [4]. Later GWAS on specific biomarkers and clinical phenotypes [5] as well as high-density genotyping in selected regions on the Immunochip [6] have yielded additional associations. A recent study used whole exome sequencing (WES) in a modest number of cases to identify specifically protein-altering variants, revealing a low-frequency variant in ATP8B4 which was enriched among SSc cases compared to controls (\(OR\ =6.1\)) [7].Of particular interest is an association from GWAS with the NOTCH4 locus which lies on chromosome 6p21 in proximity to the HLA region. This locus gave an association with the presence of anti-centromere antibody (ACA) or anti-topoisomerase I antibody (ATA) in SSc with \(P<8.84 \times 10^{-21}\), \(OR=0.55\) which were independent of the HLA class II associations [5]. The NOTCH4 locus has previously been associated, independently from the HLA, with other autoimmune disorders including ulcerative colitis [8], rheumatoid arthritis [9], and alopecia areata [10] and age-related macular degeneration [11].NOTCH4 is a member of a four-gene family (NOTCH1 to 4) and is expressed specifically in endothelial cells [12]. NOTCH proteins are transmembrane receptors activated by transmembrane ligands of the DSL family (Delta/Serrate/Lag-2). Based on structural investigation of the well-studied NOTCH1 family member, binding of the ligand triggers a conformational change in the negative regulatory region (NRR), consisting of LNR repeats and a heterodimerization (HD) region consisting of a NOD and a NODP domain (NOTCH domain) [13, 14]. The isomerization of the NRR unmasks protease cleavage sites, which leads to the intracellular domain of the NOTCH1 receptor being cleaved off. The free intracellular domain translocates to the nucleus and binds to the DNA-binding transcription factor RBP-Jk, activating transcription (Fig. 1).