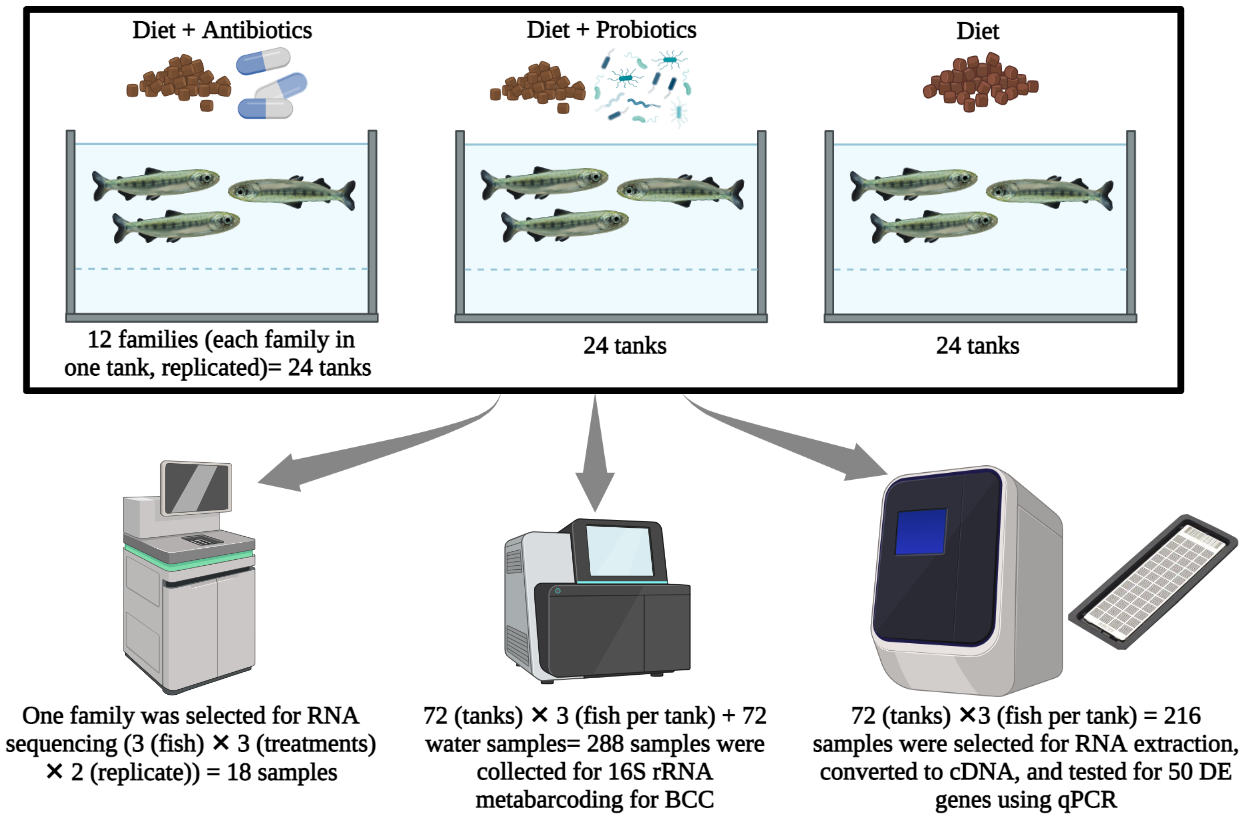
Differences in gut microbiome composition are linked with health, disease and ultimately host fitness; however, the molecular mechanisms underlying that relationship are not well characterized. Here, we modified the fish gut microbiota using antibiotic and probiotic feed treatments to address the effect of host microbiome on gene expression patterns. Chinook salmon (Oncorhynchus tshawytscha) gut gene expression was evaluated using whole transcriptome sequencing (RNA-Seq) on hindgut mucosa samples from individuals treated with antibiotic, probiotic and control diets to determine differentially expressed (DE) host genes. Fifty DE host genes were selected for further characterization using nanofluidic qPCR chips. We used 16S rRNA gene metabarcoding to characterize the rearing water and host gut microbiome bacterial communities. Daily administration of antibiotics and probiotics resulted in significant changes in fish gut and aquatic microbiota as well as more than 100 DE genes in the antibiotic and probiotic treatment fish, relative to healthy controls. Normal microbiota depletion by antibiotics mostly led to downregulation of different aspects of immunity and upregulation of apoptotic process. In the probiotic treatment, genes related to post-translation modification and inflammatory responses were up-regulated relative to controls. Our qPCR results revealed significant effects of treatment (antibiotic and probiotic) on rabep2, aifm3, manf, prmt3 gene transcription. Moreover, we found significant associations between members of Lactobacillaceae and Aeromonadaceae with host gene expression patterns. Overall, our analysis showed that the microbiota had significant impacts on many host signaling pathways, specifically targeting immune, developmental, and metabolic processes. Our characterization of some of the molecular mechanisms involved in microbiome-host interactions will help develop new strategies for preventing/ treating microbiome disruption-related diseases.