AUTHOREA
Log in
Sign Up
Browse Preprints
LOG IN
SIGN UP
Jennifer Shelton
Bioinformatics programmer
Bioinformatics Programmer at the New York Genome Center.
[email protected]
Brooklyn, NY
Member of:
New York Genome Center
Public Documents
2
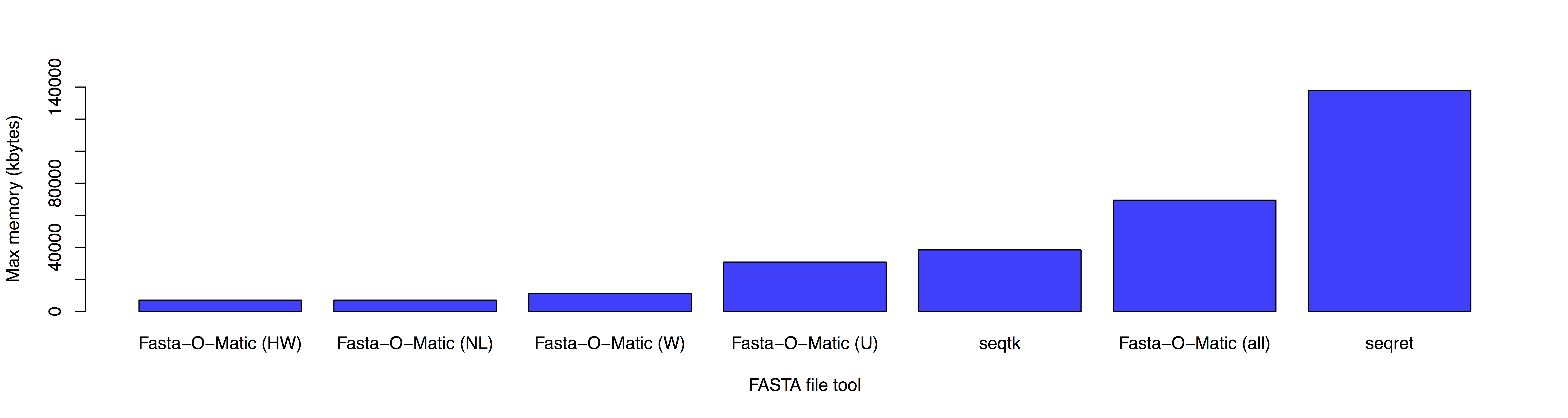
Fasta-O-Matic: a tool to sanity check and if needed reformat FASTA files
Jennifer Shelton and Sue Brown
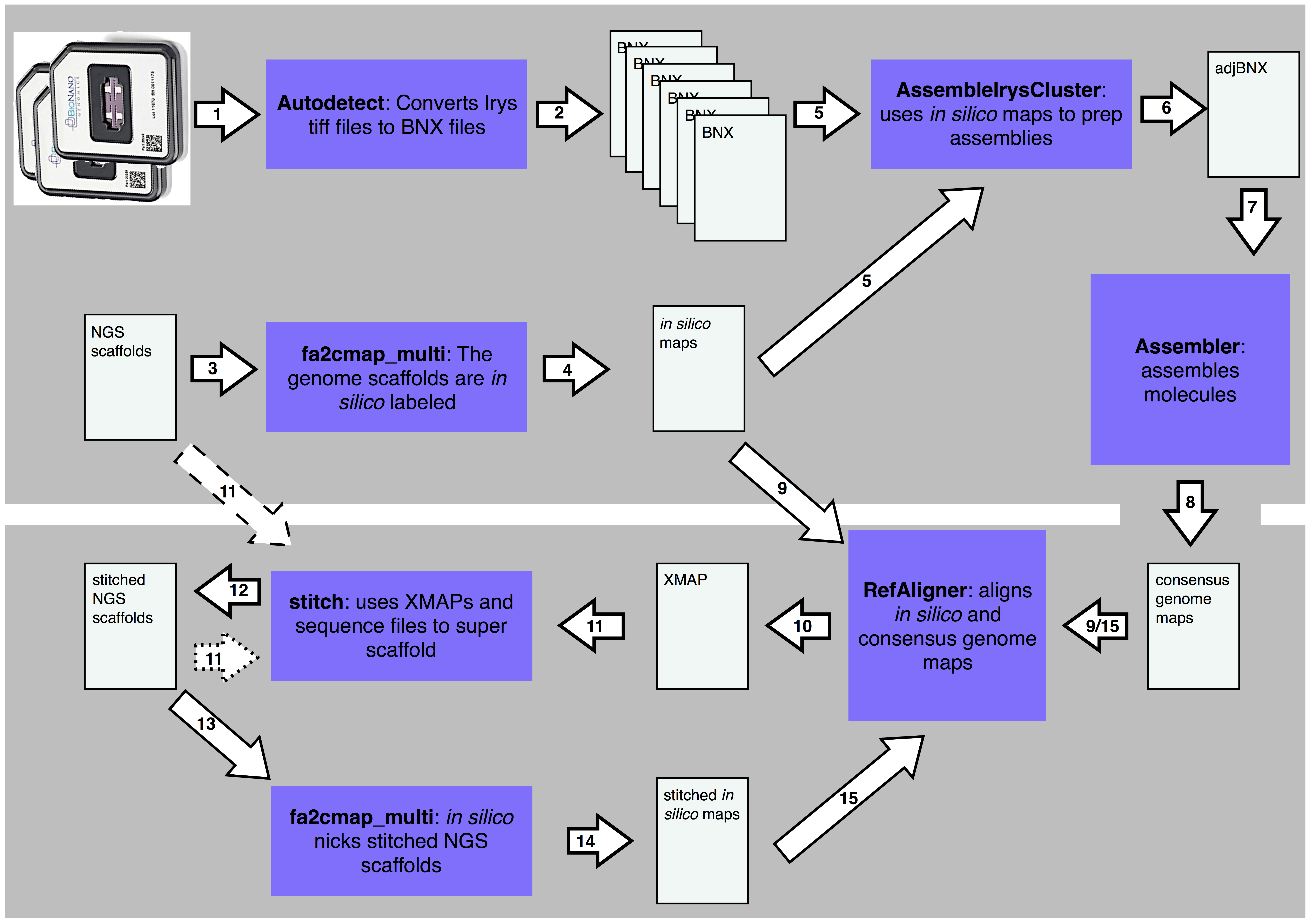
Tools and pipelines for BioNano data: molecule assembly pipeline and FASTA super scaf...
Jennifer Shelton, Cassondra Coleman, Nic Herndon, et al.