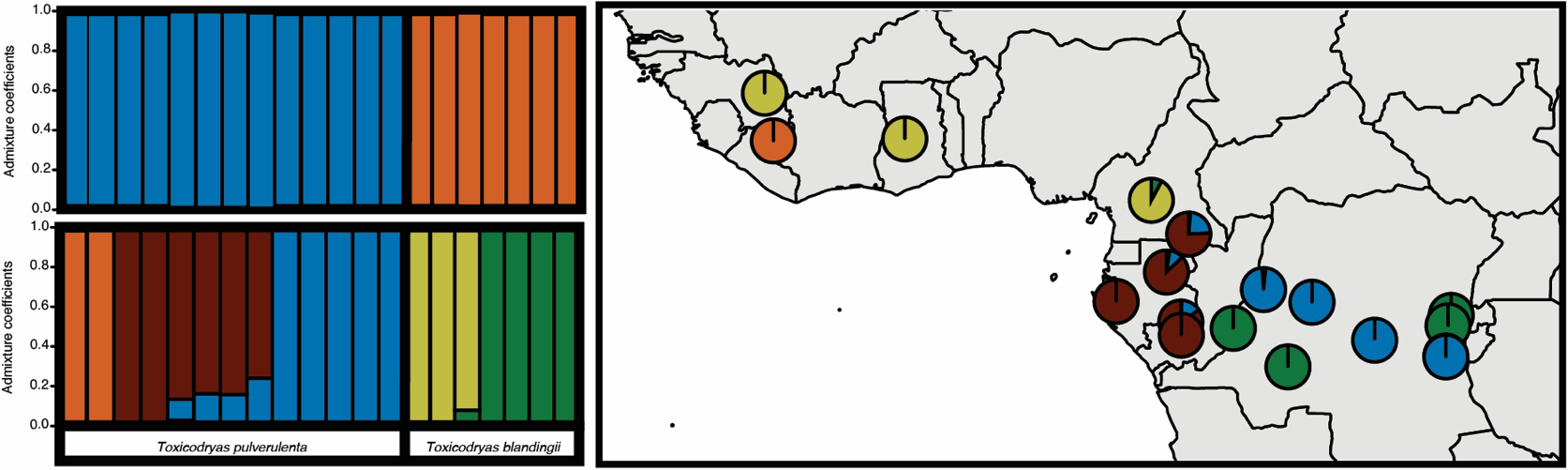
The relative roles of rivers and refugia in shaping the high levels of species diversity in tropical rainforests has been widely debated for decades. Only recently has it become possible to take an integrative approach to answer these questions with genomic sequencing and paleo-species distribution modeling. Here, we tested the predictions of the classic river, refuge, and river-refuge hypotheses on diversification in the arboreal West and Central African snake genus Toxicodryas. We used dated phylogeographic inferences, population clustering analyses, machine learning-based demographic model selection, species paleo-distribution range estimates, and climate stability modeling to conduct a comprehensive phylogenomic and historical demographic analysis of this genus. Our results revealed significant population genetic structure within both Toxicodryas species, corresponding geographically to river barriers, and divergence times ranging from the mid to late Miocene. Our demographic and migration analyses supported our interpretation that rivers have represented strong barriers to gene flow among populations since their divergence. Additionally, we found no support for a major contraction of suitable habitat during the last glacial maximum, allowing us to reject both the refuge and river-refuge hypotheses in favor of the river barrier hypothesis. This study highlights the complexity of diversification dynamics in the African tropics and the advantage of integrative approaches to studying speciation in tropical regions.