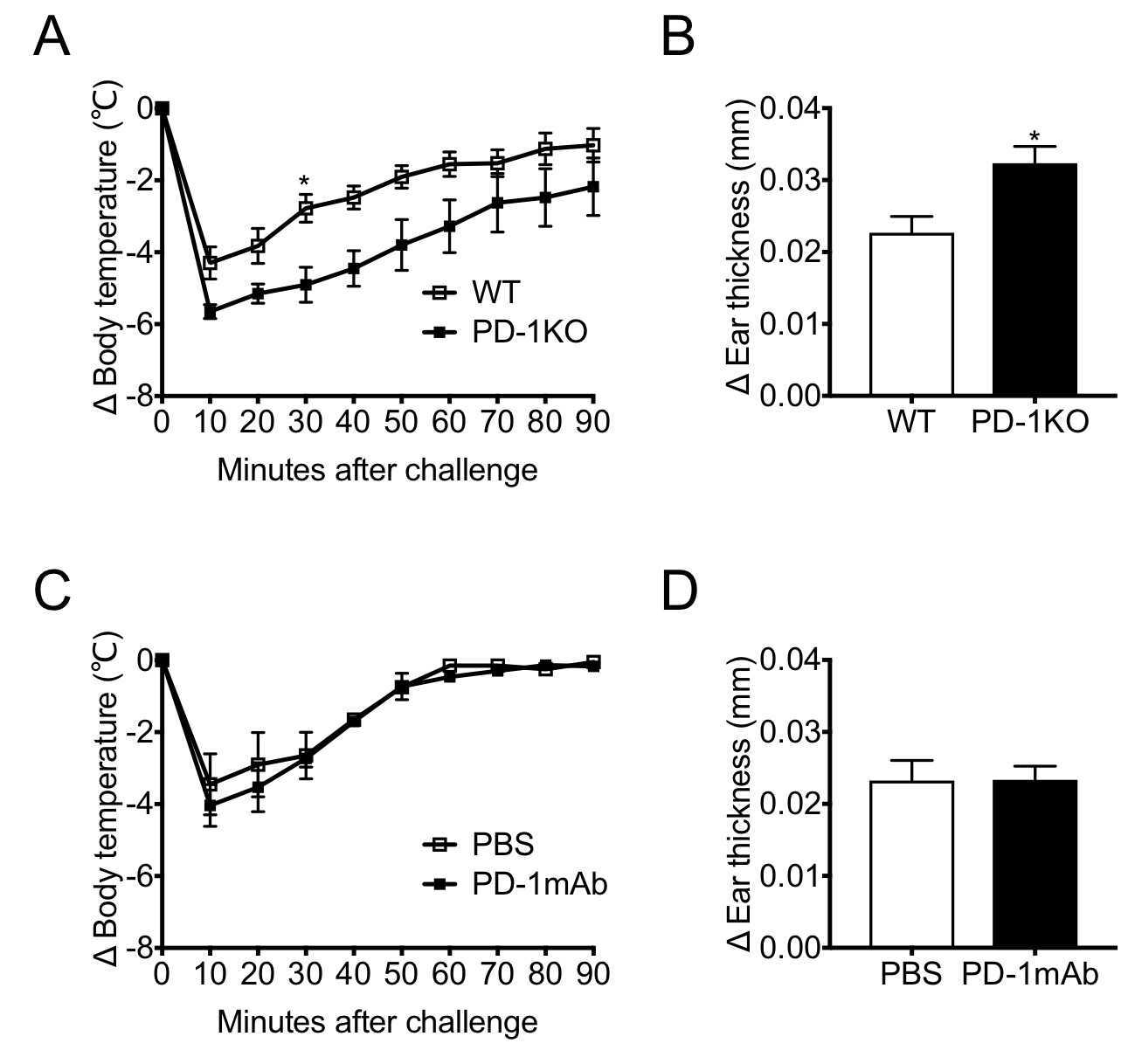
To the Editor:Antigen binding to a receptor initiates a cascade of intracellular signaling pathways and efficiently mounts an adaptive immune response in a given tissue microenvironment. Immunoreceptor engagement activates cytoplasmic protein tyrosine kinases such as SRC proto-oncogene (SRC), LYN proto-oncogene (LYN), and spleen associated tyrosine kinase. These kinases phosphorylate the immunoreceptor tyrosine-based activation motif (ITAM) and activate downstream immune effector functions through their interaction with the cytoplasmic tail. However, excessive activation can result in immune-mediated tissue damage. The cytoplasmic tail of programmed cell death-1 (PD-1) harbors an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM).1 Like ITAM, these highly conserved motifs are phosphorylated upon immunoreceptor engagement2 and interact with protein tyrosine phosphatase non-receptor type 6 or protein tyrosine phosphatase non-receptor type 11 (SHP-2). It is known that PD-1 interacts with SHP-2 and regulates antigen receptor signaling pathways through the cytoplasmic tail.3, 4The mast cell (MC) is not only a key component of innate immunity but also serves as a critical effector in the adaptive immune response. Analogous to the T/B cell receptor signaling pathways, the high-affinity immunoglobin E receptor (FcεRI) signaling pathway involves ITAM phosphorylation, which in turn results in degranulation and the release of mediators/enzymes. Conversely, the ITIM/ITSM phosphorylation event negatively regulates the MC effector function.5Although it was recently shown that the ligation of PD-1 on MCs induces peripheral tolerance,6 there has been a paucity of direct evidence showing the inhibitory role of PD-1 on MC effector function in vivo . To this end, we employed the IgE-mediated passive anaphylaxis model. We compared the consequences of total PD-1 absence with PD-1 receptor blockade7 and delineated the requirement of the cytoplasmic tail for the regulation of FcεRI signaling.In contrast to the findings for MC-specific deletion of SHP-2,8 cutaneous, or peritoneal MC populations in PD-1 knockout (PD-1KO) mice were not altered significantly (Fig. S1a, b). This discrepancy suggests that PD-1 is dispensable for the development of the tissue MC lineage on the KIT proto-oncogene/SHP-2 axis.8 Regarding the passive systemic anaphylaxis, PD-1KO mice exhibited a significantly increased drop in body temperature compared with wild-type (WT) mice (Fig. 1a). Likewise, in the late-phase passive cutaneous anaphylaxis, PD-1KO mice showed a significantly enhanced ear swelling compared with WT mice (Fig. 1b). However, a monoclonal antibody (mAb)-mediated PD-1 receptor blockade did not exacerbate the anaphylactic response (Fig. 1c, d, and Fig. 2).Monoclonal antibodies targeting the PD-1/programmed death-ligand 1 (PD-L1) pathway or cytotoxic T-lymphocyte antigen 4 have revolutionized the medical oncology. Nonetheless, such a therapeutic approach inherently runs the risk of evoking immune-related adverse events (irAEs).9 Previously, we have shown that either genetic deletion of PD-1 or mAb-mediated PD-1 receptor blockade exacerbates allergic contact dermatitis, and we inferred that the PD-1/PD-L1 pathway is a critical regulator of cutaneous irAE (C-irAE).7 In contrast to the cell-mediated cutaneous immune response, we found that the augmented IgE humoral immune response required the total absence of PD-1, but immunoreceptor engagement was dispensable. This discrepancy may be analogous to the fact that the cytoplasmic PD-1 tail can interact with the SHP-2 phosphatase upon T cell receptor ligation, even in the absence of PD-1 receptor engagement.2 These lines of evidence and our results further suggest that PD-1 evolved to regulate the adaptive immune response at the effector phase7 along with other inhibitory immunoreceptors. Our results may also correlate with clinical observations. Although the urticarial rash is the most common form of C-irAE, anaphylaxis, the systemic counterpart of urticaria has never been reported in this context. Therefore, our results could be an important guide for the medical oncology practice in that aggravated IgE-mediated anaphylaxis is due to the total absence of PD-1, but not receptor engagement.