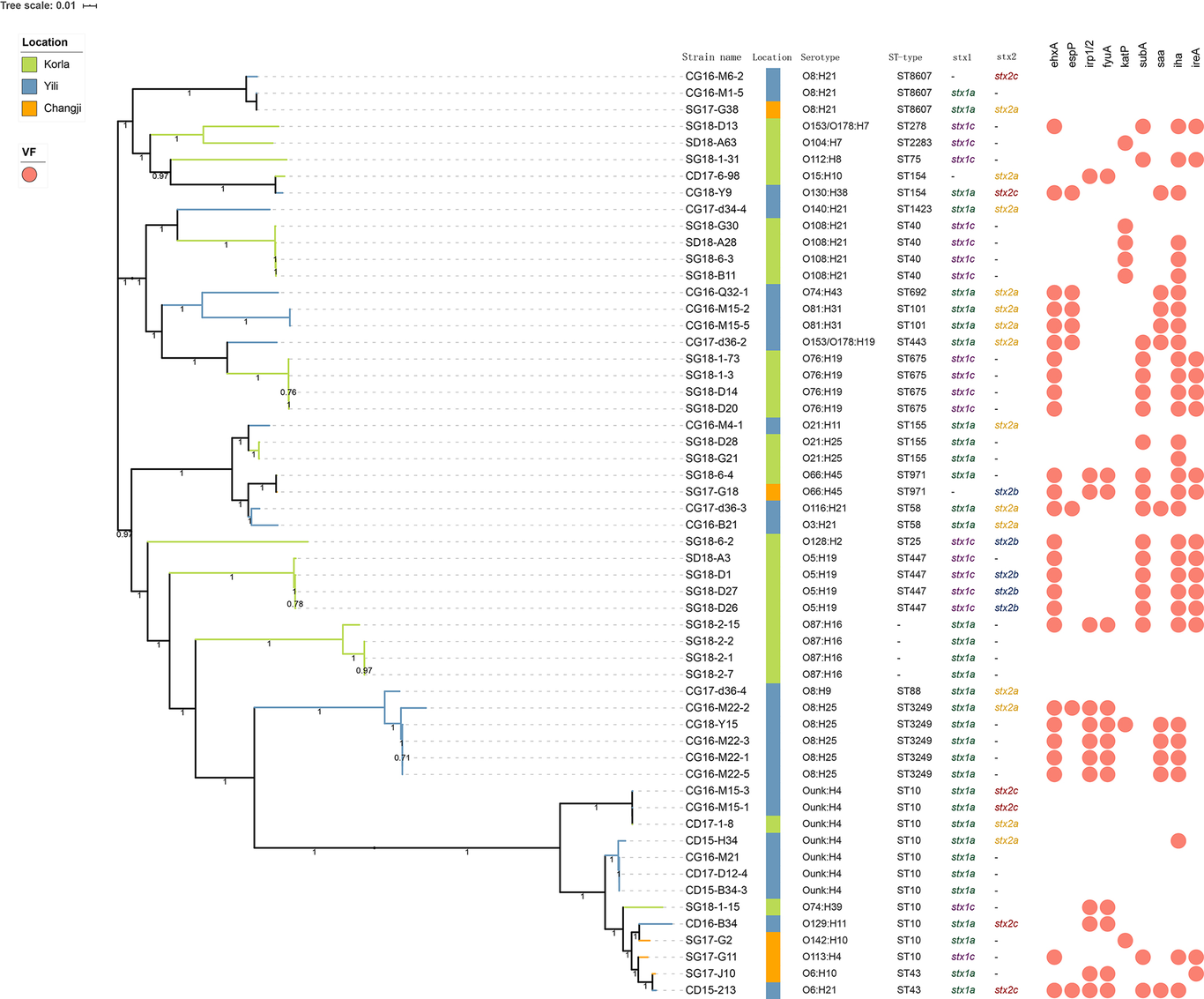
Shiga toxin-producing Escherichia coli (STEC) is an important foodborne pathogen capable of causing severe gastrointestinal diseases in humans. Cattle and sheep are the natural reservoir hosts of STEC strains. Previously, we isolated 56 STEC strains from anal and carcass swab samples of cattle and sheep in farms and slaughterhouses. In this study, we performed whole-genome sequencing of these isolates and determined their serotypes, virulence profiles, sequence types (STs), and genetic relationships. Our results showed that the 56 isolates belong to 20 different STs, 29 O:H serotypes, and 8 stx subtype combinations. The highly prevalent serotypes were O8:H25 and O87:H16 for bovine and ovine isolates, respectively. Five serotypes of cattle or sheep isolates are novel. The majority (63%) of cattle isolates contain stx1+stx2, subtyped into stx1a, stx2a, and stx2c. In contrast, most of the sheep isolates contain stx1 only, primarily subtyped into stx1a and stx1c. None of the isolates tested eae-positive, but virulence factors such as ehxA and espP were present with variable prevalence rates. The prevalence of saa (19.6%) and espP (12.5%) in cattle isolates is much higher than that in sheep isolates, whereas that of subA (34%), katP (14.3%), and ireA (28.6%) in sheep isolates is considerably higher than that in cattle isolates. Core-genome SNP analysis revealed that the majority of isolates could be clustered based on their serotypes or STs, whereas some clustering is associated with more than one ST or serotype. Seven-gene Multilocus Sequence Typing (MLST) indicated that nine sheep isolates and four cattle isolates were related to a few E. coli isolates associated with human HUS, suggesting their potential in causing severe human infections. Collectively, we described the characteristics of cattle and sheep STEC isolates from Xinjiang, China, which may be utilized in comparative studies of other geographic regions and sources of isolation and for surveillance.