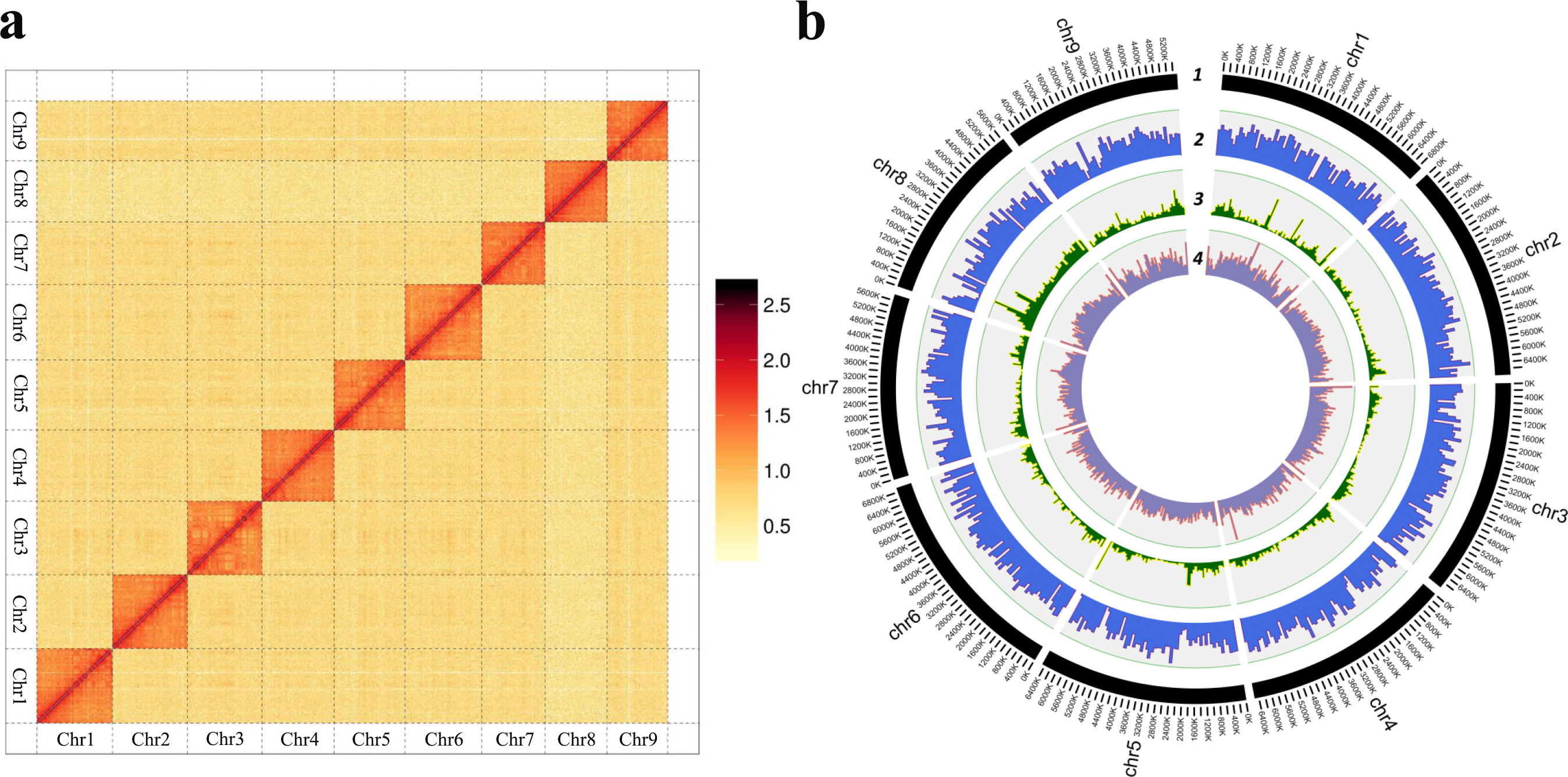
Background: Mites represent the second largest group with diverse niches and feeding habits, except for insects. Scabies mites are the causative agents of highly contagious skin disease in humans and more than 100 mammals. Although several versions of Sarcoptes scabiei genome have been published, i.e. var. suis, var. canis and var. hominis, the chromosome-level genome and population divergence is still desired for the community. Besides, the molecular mechanisms that scabies mites adapt to a parasitic lifestyle remains unclear. The taxonomy and ancestral origin of the scabies mite is unknown. Results: Here, we reported the first chromosome-level reference genome of S. scabiei, which was isolated from rabbits. The genome has a contig N50 size of 5.92 Mb, a total assembled length of 57.30 Mb, and ~12.65% of repetitive sequences and 9,333 protein‑coding genes were predicted. Population genetics analysis supported that scabies mites isolated from different hosts can be subdivided by hosts, and humans are likely the primary hosts of scabies mites, followed by pigs, dogs, and rabbits. However, phylogeny results suggested that rabbit was infected with scabies long before they were domesticated by humans, contradicting previous hypothesis that humans transmitted scabies mites to animals through domestication. Comparative genomics between scabies mites and mites of other feeding habits provided clues concerning the mechanisms of adaptation to permanent parasitic life from morphology, detoxification, and metabolism. Conclusions: Together, the first chromosome-level S. scabiei genome and population genetics analysis indicated its genetic subdivisions and within-host species divergence, which also provide evidence for further control of this highly contagious skin disease.