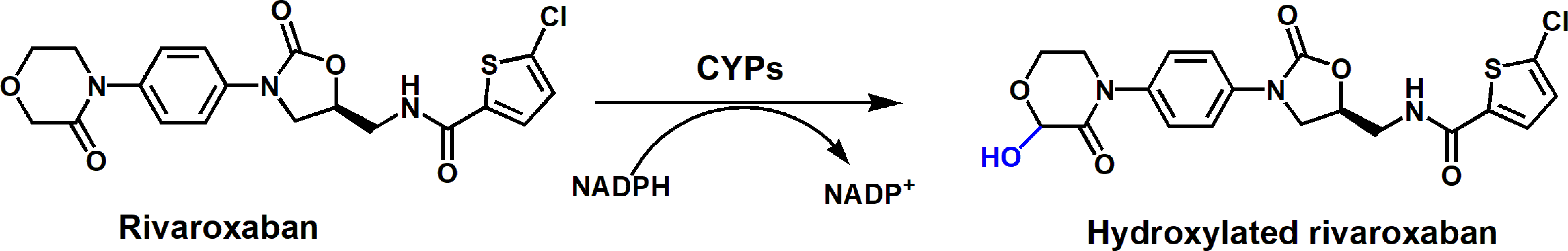
Aim Rivaroxaban, an oral anticoagulant, undergoes the metabolism mediated by human cytochrome P450 (CYP). The present study is to quantitatively analyze and compare the contributions of multiple CYPs in the metabolism of rivaroxaban to provide new information for medication safety. Methods The metabolic stability of rivaroxaban in the presence of human liver microsomes (HLMs) and recombinant CYPs was systematically evaluated to estimate the participation of various CYP isoforms. Furthermore, the catalytic efficiency of CYP isoforms was compared via metabolic kinetic studies of rivaroxaban with recombinant CYP isoenzymes, as well as via CYP-specific inhibitory studies. Additionally, docking simulations were used to illustrate molecular interactions. Results Multiple CYP isoforms were involved in the hydroxylation of rivaroxaban, with decreasing catalytic rates as follows: CYP2J2 > 3A4 > 2D6 > 4F3 > 1A1 > 3A5 > 3A7 > 2A6 > 2E1 > 2C9 > 2C19. Among the CYPs, 2J2, 3A4, 2D6 and 4F3 were the four major isoforms responsible for rivaroxaban metabolism. Notably, the intrinsic clearance of rivaroxaban catalyzed by CYP2J2 was nearly 39-, 64- and 100-fold that catalyzed by CYP3A4, 2D6 and 4F3, respectively. In addition, rivaroxaban hydroxylation was inhibited by 41.1% in the presence of the CYP2J2-specific inhibitor danazol, which was comparable to the inhibition rate of 43.3% by the CYP3A-specific inhibitor ketoconazole in mixed HLMs. Furthermore, molecular simulations showed that rivaroxaban principally bound to CYP2J2 by π-alkyl bonds, carbon-hydrogen bonds and alkyl interactions. Conclusion CYP2J2 dominated the hydroxylation of rivaroxaban, which may provide new insight into clinical drug interactions involving rivaroxaban.