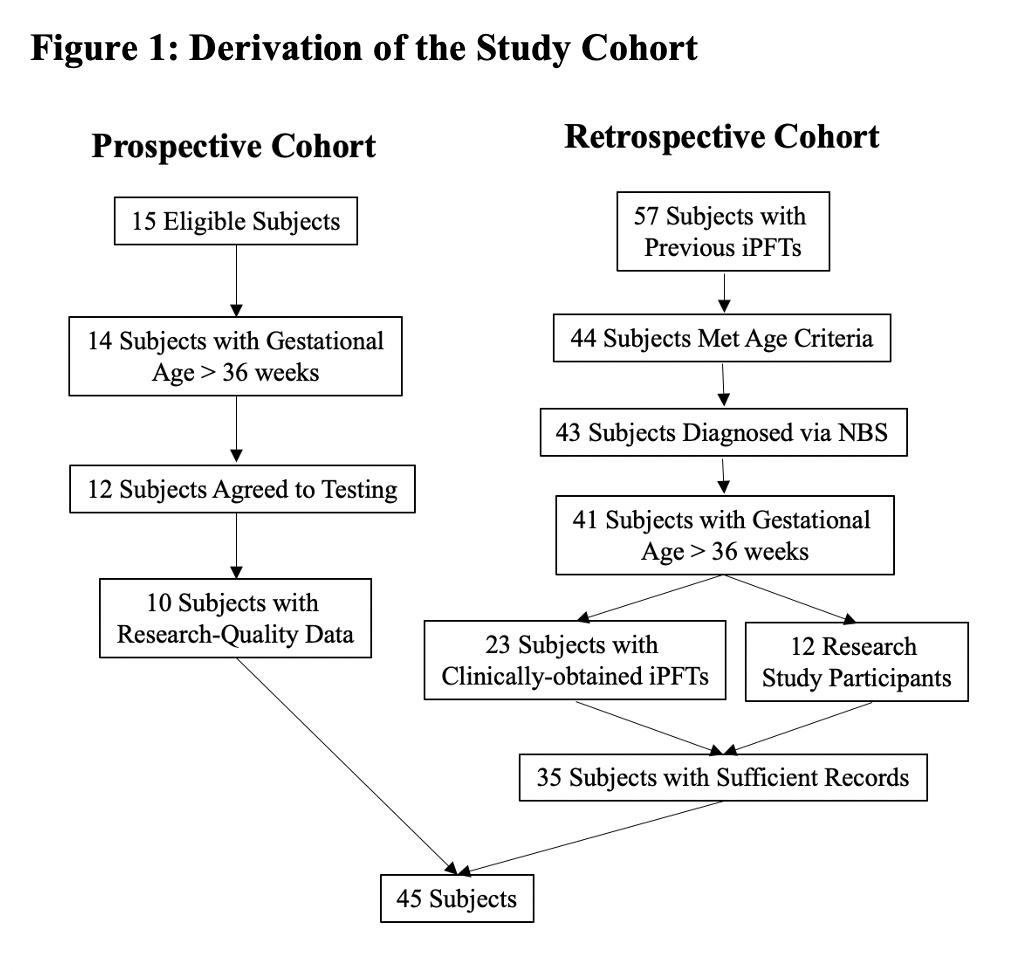
ABSTRACT Background: The goal of this study was to identify clinical features associated with abnormal infant pulmonary function tests (iPFTs), specifically functional residual capacity (FRC), in infants with cystic fibrosis (CF) diagnosed via newborn screen (NBS). We hypothesized that poor nutritional status in the first 6-12 months would be associated with increased FRC at 12-24 months. Methods: This study utilized a combination of retrospectively and prospectively collected data from ongoing research studies and iPFTs performed for clinical indications. Demographic and clinical features were obtained from the electronic medical record. Forced expiratory flows and volumes were obtained using the raised volume rapid thoracoabdominal technique (RVRTC) and FRC was measured via plethysmography. Results: A total of 45 CF NBS infants had iPFTs performed between 12-24 months. Mean forced vital capacity, forced expiratory volume in 0.5 second, and forced expiratory flows were all within normal limits. In contrast, the mean FRC z-score was 2.18 (95%CI=1.48, 2.88) and the mean respiratory rate (RR) z-score was 1.42 (95%CI=0.95, 1.89). There was no significant association between poor nutritional status and abnormal lung function. However, there was a significant association between higher RR and increased FRC, and a RR cutoff of 36 breaths/min resulted in 92% sensitivity to detect hyperinflation with 32% specificity. Conclusions: These results suggest that FRC is a more sensitive measure of early CF lung disease than RVRTC measurements and that RR may be a simple, non-invasive clinical marker to identify CF NBS infants with hyperinflation.