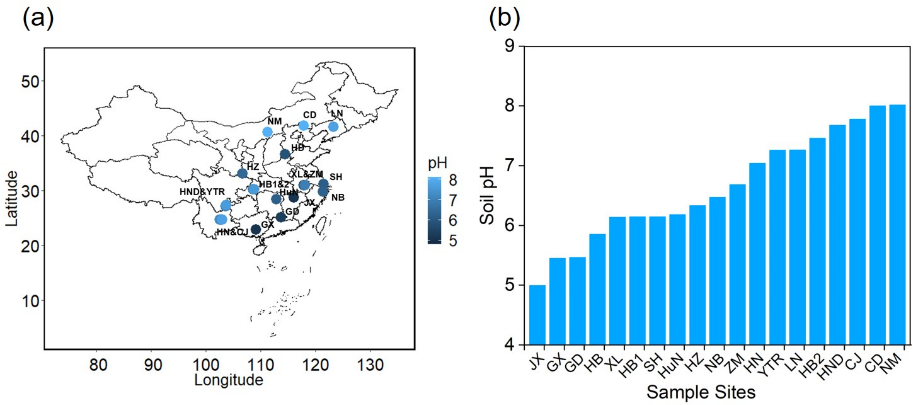
Increasing evidence suggests that fungal communities are key components of biogeochemical cycles in coastal ecosystems. While several studies highlighted strong spatial patterns in fungal abundance and diversity, there are very few studies using a more integrative approach to study the spatio-temporal distribution of fungi, taking also the active part of the community into account. To better understand the consequences of anthropogenic activities, e.g. marine aquaculture, for fungal community composition and activities, we simultaneously evaluated the temporal (four different seasons) and spatial dynamics in total (DNA) and active (RNA) fungal communities in relation to several major physicochemical properties. Fungal communities were highly diverse, but showed the ubiquitous dominance of Dikarya and the occasional predominance of Glomeromycota, Mucoromycota, Mortierellomycota, Chytridiomycota, Mortierellomycota, Olpidiomycota, and Rozellomycota. Thereby, fungal diversity indices showed a much higher seasonal variation than with the degree of aquaculture activity, for both total and active communities. This notion is supported by co-occurrence networks exhibiting a clear seasonal pattern. Furthermore, fungal community structure in coastal waters showed distinct relationships with environmental factors varying both with season and in space. For both, total and active fungal communities, a combination of environmental variables such as temperature, DO and NO2- exhibited the greatest impact on community structure. Our study demonstrates a distinct spatio-temporal dynamics of both, total and active fungi and provides a foundation to better understand the ecological roles of marine fungi in coastal ecosystems in relation to mariculture activities.