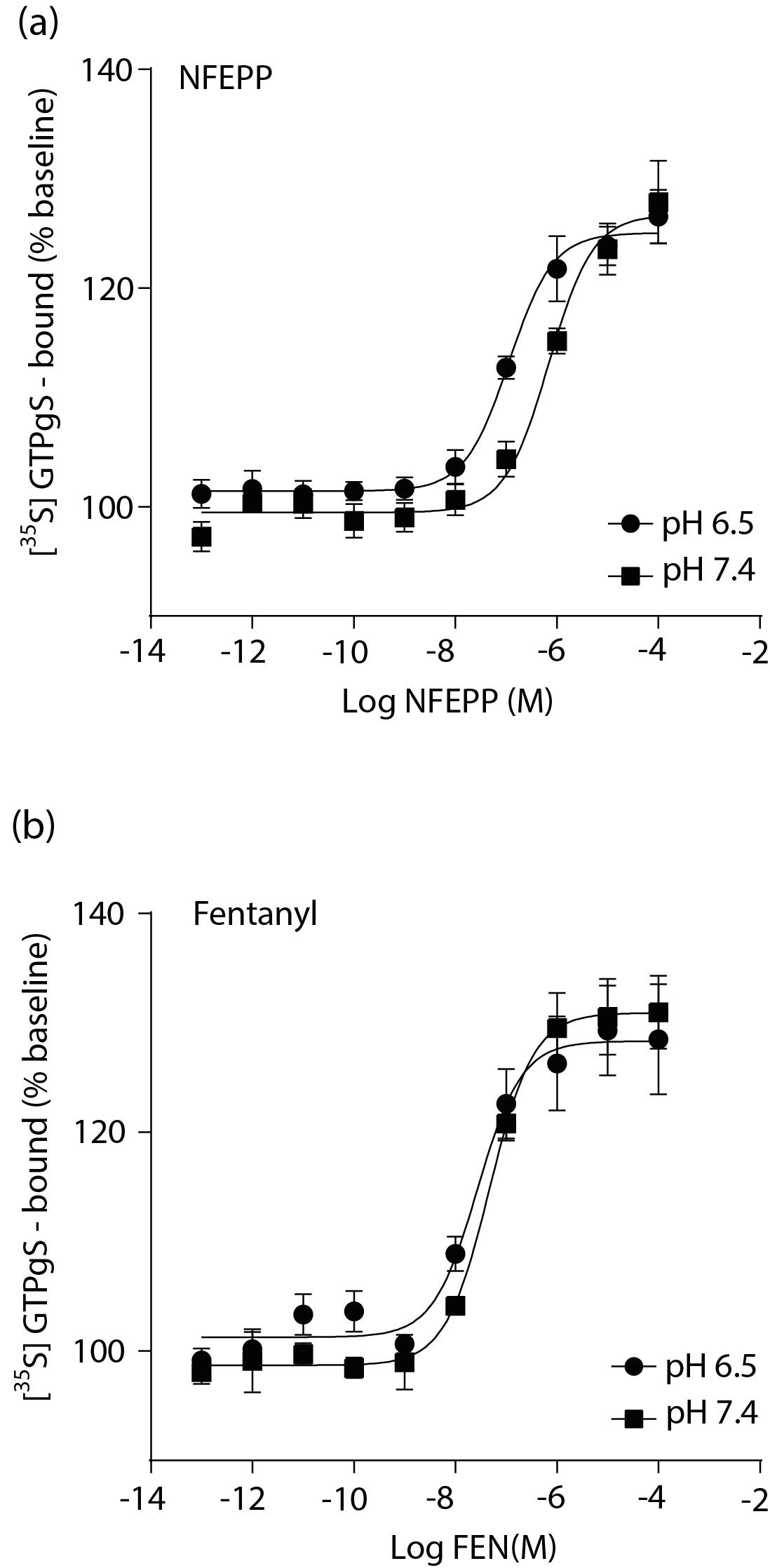
Abstract Background and Purpose NFEPP is a newly-designed pain killer selectively activating G-protein coupled mu opioid receptors in injured tissues, and therefore devoid of central side effects. However, the cellular mechanisms underlying NFEPP’s antinociceptive effects were not examined in sufficient detail so far. Here we investigated the effects of NFEPP on G-protein activation, on voltage gated calcium channels and on mu opioid receptor phosphorylation. Experimental Approach HEK293 cells stably transfected with mu opioid receptors were used to study [35S]-GTPγS binding and mu opioid receptor phosphorylation. Voltage dependent calcium currents and intracellular calcium signals were examined in rat sensory neurons. All experiments were performed at acidic and physiological pH values using NFEPP compared to the conventional mu opioid receptor agonist fentanyl. To investigate the role of G protein subunits, we used pertussis toxin and gallein. Key Results At low pH, NFEPP produced more efficient G-protein activation and reduction of calcium currents in depolarized sensory neurons. The latter was mediated by G protein βγ subunits and NFEPP-mediated MOR phosphorylation was pH-dependent. Fentanyl-induced signaling was not affected by pH changes. Conclusion and Implications Our study shows that, at low pH, MOR signaling induced by NFEPP is more effective and neuronal calcium channels are directly modulated by G protein βγ subunits dissociated from G protein αi/o subunits. Apparently, the enhanced efficacy of NFEPP is dependent on extra- rather than intracellular effects on opioid receptor function.