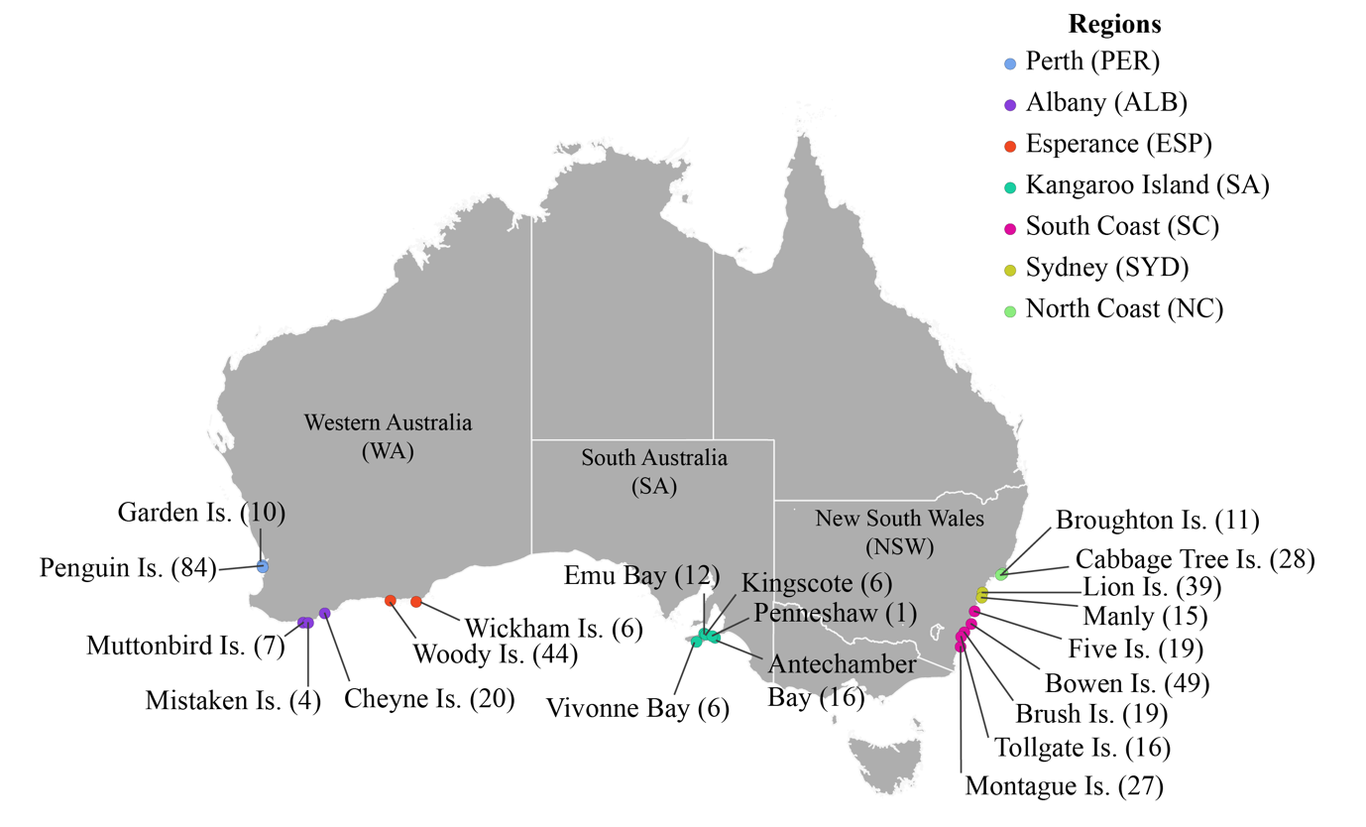
The Australian range of little penguins, Eudyptula minor, extends around southern Australia, with range-edge sites near the large cities of Perth (west) and Sydney (east). Both range-edges are closer to the equator than the range-core, being likely to experience similar heating with climate change. As a result, movement to one range-edge is not an option for little penguins, unlike in many other species. Therefore, adaptation at the range edge might be very important for little penguins. Capacity for future adaptation depends upon the variability each site holds, and the amount of exchange between sites. In peripheral sites, incoming dispersal might either forestall demographic collapse and replenish genetic variation (good), or overcome local adaptation and increase disease transmission (bad). We aimed to establish the genetic variability in each site, and the exchange (dispersal) of individuals between sites. Genetic markers included biparentally-inherited microsatellites, and maternally-inherited mitochondrial DNA sequence. For microsatellites, no site appeared to have critically low variation, including the peripheral sites, however there was a significant but slight trend of increased variation from east to west. In contrast, mitochondrial DNA showed a pattern of significantly reduced variation at the two range-edges, possibly indicating differential dispersal patterns in males and females. There appear to be two main genetically distinct groups, in the west and the east, but analysis of lifetime dispersal patterns across the Australian range also suggests complex dispersal, sometimes with high dispersal or similarity between locations that are not adjacent. Our work suggests that despite some differentiation, little penguin sites are interdependent due to complex dispersal patterns, and all have valuable genetic variation. In particular, the peripheral sites are not depauperate of variation, and are moderately connected to the remainder of the distribution, so possibly may be able to adapt in response to climate warming.