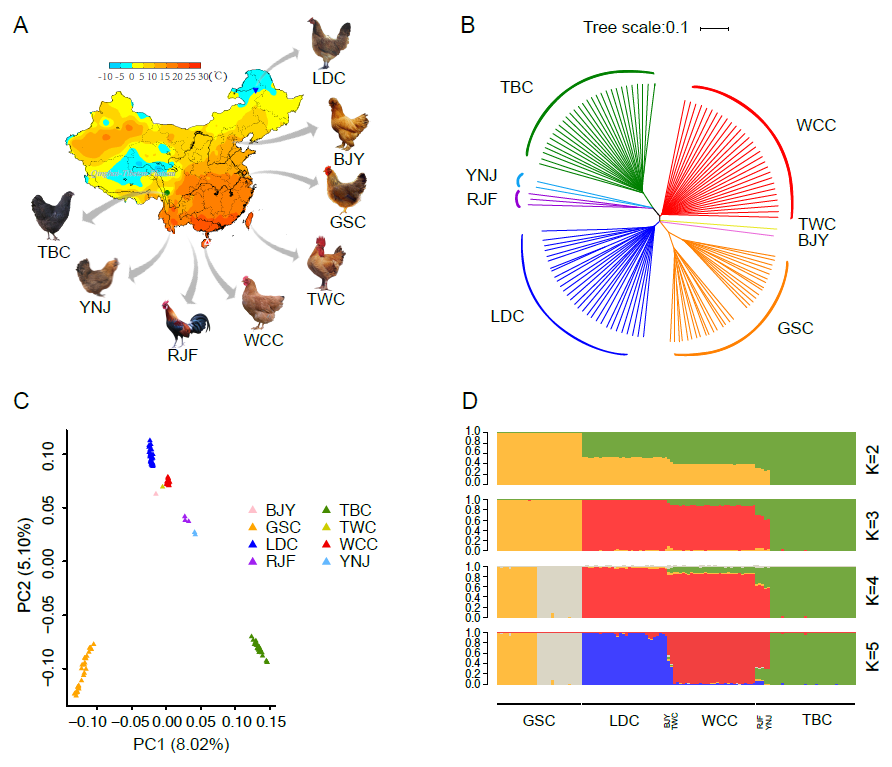
Investigating the genetic footprints of historical temperature selection can get insights to local adaptation and potential influences of climate change on long-term population dynamics. Chicken is a significative species to study genetic adaptation on account of its similar domestication track related to human activity with the most diversified varieties. Yet, few studies have demonstrated the genetic signatures of its adaptation to naturally tropical and frigid environments. Here, we generated whole genome resequencing of 119 domesticated chickens in China including the following breeds which are in order of breeding environmental temperature from more tropical to more frigid: Wenchang chicken (WCC), green-shell chicken (GSC), Tibetan chicken (TBC), and Lindian chicken (LDC). Our results showed WCC branched off earlier than LDC with an evident genetic admixture between WCC and LDC, suggesting their closer genetic relationship. Further comparative genomic analyses SLC33A1 and TSHR genes exhibited stronger signatures for positive selection in the genome of the more tropical WCC. Furthermore, genotype data from about 3,000 African local ecotypes confirmed that allele frequencies of SNPs in these two genes appeared strongly associated with tropical environment adaptation. In addition, the NDUFS4 gene exhibited a strong signature for positive selection in the LDC genome, and SNPs with marked allele frequency differences indicated a significant relationship with frigid environment adaptation. Our findings partially unravel how selection footprints from environmental temperature stress can lead to advantageous genomic adaptions to tropical and frigid environments in poultry and provides a valuable resource for selective breeding of chickens and other domestic animals.